

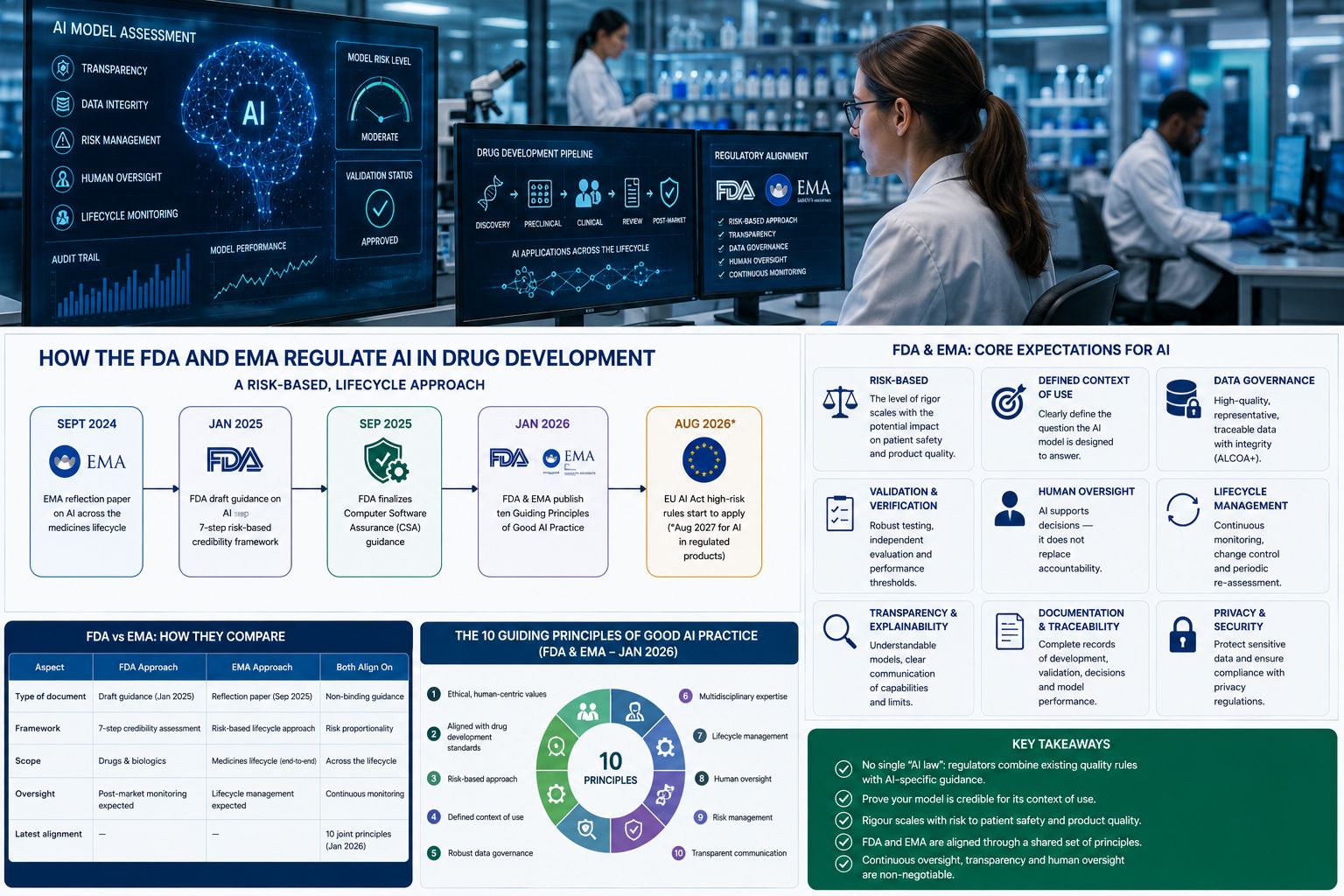
The FDA and EMA regulate AI in drug development through a risk-based, lifecycle approach: AI used to help decide a medicine’s safety, efficacy or quality must be transparent, validated and demonstrably fit for its specific purpose. The FDA set this out in draft guidance in January 2025, the EMA in a reflection paper in September 2024, and in January 2026 the two agencies jointly published ten “Good AI Practice” principles to align their expectations.
TL;DR: There is no single “AI law” for drug development. Instead, the FDA and EMA apply existing quality rules plus new AI-specific guidance built on one idea — prove your model is credible for how you actually use it. Requirements scale with risk, oversight is continuous, and US and EU expectations are converging fast.
Who should read this? This guide is for regulatory affairs and quality professionals, clinical and biostatistics teams, drug developers and sponsors, AI/ML and data-science leads in pharma and biotech, and vendors supplying AI into regulated drug development.
AI now touches every stage of the medicines lifecycle, from target discovery to pharmacovigilance. But if an AI model informs a regulatory decision, regulators expect evidence that it can be trusted. This guide explains what the FDA, EMA and other authorities actually require, how their frameworks compare, and what companies need to do to deploy AI that will pass regulatory scrutiny.
Quick Navigation
- What are the rules for AI in drug development?
- The FDA approach
- The EMA approach
- FDA–EMA harmonisation: the ten principles
- The EU AI Act and other rules
- FDA vs EMA: how they compare
- What companies need to do
- Business impact and industry implications
- Challenges and opportunities
- Future Outlook & LSGN Perspective
- FAQs
At a Glance: Key Statistics
- September 2024 — the EMA finalised its reflection paper on AI across the medicines lifecycle.
- January 2025 — the FDA issued its first draft guidance on AI to support regulatory decisions, with a 7-step risk-based credibility framework (final expected 2026).
- September 2025 — the FDA finalised its risk-based Computer Software Assurance guidance.
- January 2026 — the FDA and EMA jointly published ten Guiding Principles of Good AI Practice in Drug Development.
- 2 August 2026 — the EU AI Act’s high-risk rules begin to apply (extended to 2 August 2027 for AI embedded in regulated products).
Figures are drawn from the regulatory sources linked above; verify before publishing.
Key takeaways
- There is no standalone “AI statute” for drug development — regulators combine existing quality rules with new AI-specific guidance.
- The shared core is credibility for a defined context of use: prove the model is fit for the exact decision it informs, with rigour proportionate to risk.
- The FDA (draft AI guidance + CSA) and EMA (reflection paper) have converged, formalised by their January 2026 joint principles.
- In the EU, the EU AI Act adds a separate, horizontal layer — especially for AI that is or sits inside a medical device.
- Human oversight, data governance, transparency and lifecycle monitoring are non-negotiable across every framework.
What are the rules for AI in drug development?
Unlike some sectors, life sciences has no single, dedicated “AI in medicines” law. Instead, AI is governed by a layered system: long-standing quality and records rules (such as GxP, 21 CFR Part 11 and EU GMP Annex 11), plus newer AI-specific guidance from the FDA and EMA, plus — in the EU — the horizontal EU AI Act. What ties them together is a single expectation: if AI output supports a regulated decision, the company must be able to demonstrate the model is credible, controlled and fit for its purpose.
Key takeaway: Regulators do not ask “is your AI good?” — they ask “have you proven it is trustworthy for this specific use?”
The FDA approach
In January 2025 the FDA released its first draft guidance on using AI to support regulatory decision-making for drugs and biologics. Its centrepiece is a seven-step, risk-based credibility-assessment framework organised around the model’s context of use:
- Define the question of interest.
- Define the AI model’s context of use.
- Assess the AI model risk.
- Develop a credibility-assessment plan.
- Execute the plan.
- Document the results and deviations.
- Determine adequacy and manage the model over its life cycle.
The public comment period closed in April 2025 and final guidance is expected in 2026. This sits alongside the FDA’s finalised Computer Software Assurance guidance (2025) and long-standing rules such as 21 CFR Part 11.
The EMA approach
The EMA finalised its reflection paper on the use of AI in the medicinal product lifecycle in September 2024, adopted by both the CHMP and CVMP. It is not binding law but sets out the agency’s thinking across discovery, non-clinical and clinical development, regulatory review, manufacturing and post-authorisation. Like the FDA, the EMA champions a risk-based approach, with developers expected to proactively identify risks and manage AI/ML tools across their full lifecycle. Companies can also seek EMA scientific advice or qualification for specific AI methodologies.
FDA–EMA harmonisation: the ten principles
In January 2026 the FDA and EMA took a significant step toward alignment, jointly publishing ten Guiding Principles of Good AI Practice in Drug Development. In essence, the principles call for: alignment with ethical, human-centric values; adherence to established drug-development standards; a risk-based approach; a clearly defined context of use; robust data governance; multidisciplinary expertise; lifecycle management; human oversight; risk management; and clear, accessible communication about AI systems.
Key takeaway: A company that builds to the ten joint principles is, in practice, building for both markets at once.
The EU AI Act and other rules
In the EU, the EU AI Act adds a separate, horizontal layer of regulation on top of medicines law. It classifies AI systems by risk; crucially, AI that is — or is embedded in — a medical device or IVD is automatically treated as high-risk, triggering strict obligations. Its high-risk rules begin to apply from 2 August 2026, with an extended transition to 2 August 2027 for AI embedded in regulated products. In the UK, the MHRA runs its own “Software and AI as a Medical Device” programme and AI-focused regulatory sandbox, and the international ICH is expected to shape future global harmonisation.
FDA vs EMA: how they compare
| Dimension | FDA (US) | EMA (EU) |
|---|---|---|
| Flagship AI document | Draft AI guidance (Jan 2025) | Reflection paper (Sept 2024) |
| Core method | 7-step risk-based credibility assessment | Risk-based, lifecycle approach |
| Status | Draft; final expected 2026 | Finalised (non-binding) |
| Supporting rules | 21 CFR Part 11; CSA (2025) | EU GMP Annex 11; EU AI Act |
| Engagement route | Formal FDA interactions / meetings | Scientific advice; methodology qualification |
| Shared foundation | Ten Guiding Principles of Good AI Practice (joint, Jan 2026) | |
Table: the frameworks differ in form and status, but their underlying philosophy — risk-based credibility for a defined context of use — is now shared.
What companies need to do
- Define context of use first. Pin down exactly what decision the model informs and its regulatory weight before building.
- Assess and document risk. Match the depth of your evidence to the patient and product impact.
- Govern your data. Evidence quality, representativeness, bias assessment and integrity (ALCOA+).
- Keep humans in the loop. Define human oversight for high-impact decisions.
- Plan for the lifecycle. Monitor performance and drift; set triggers for revalidation and change control.
- Engage regulators early. Use FDA meetings or EMA scientific advice to de-risk novel approaches.
Business impact and industry implications
Regulatory clarity is arriving faster than many expected, and that is broadly good news: defined expectations let companies invest in AI with more confidence and fewer nasty surprises at submission. But it also raises the bar. Regulatory affairs and data science must now work as one team, and “credibility evidence” becomes a deliverable planned from day one, not bolted on at the end. For vendors, regulator-ready AI is becoming a selling point — the same shift LSGN has tracked in coverage such as Kivo’s GxP-compliant AI architecture.
Industry implication: expect regulatory-affairs teams to grow AI expertise fast, a premium on transparent and explainable models, and firms that master the credibility framework to enjoy shorter, smoother review cycles.
Challenges and opportunities
- Fragmentation. Overlapping rules (drug guidance, CSA, EU AI Act) are complex to navigate. Opportunity: the joint principles reduce duplication for firms operating in both markets.
- Uncertainty. Key texts are still draft or awaiting Commission guidelines. Opportunity: early engagement shapes and de-risks your position.
- Evidence burden. Credibility assessments take time and skill. Opportunity: reusable validation frameworks compound in value.
- Explainability. Opaque models are harder to defend. Opportunity: transparent AI wins regulatory trust faster.
Future Outlook & LSGN Perspective
Over the next three to five years, expect the FDA to finalise its AI guidance, the EU AI Act’s obligations to bite, and further international convergence — likely through the ICH. The direction is unmistakable: risk-based, lifecycle-oriented, human-centred oversight, increasingly harmonised across major markets.
Key takeaway: The regulatory question is shifting from “will regulators allow AI?” to “can you prove your AI is credible?” — and the companies ready to answer will move fastest.
LSGN Perspective: we read the January 2026 joint principles as the moment AI regulation in drug development matured from open question to working framework. The competitive edge will belong to organisations that treat credibility evidence as a core R&D deliverable and that engage regulators early rather than presenting finished models and hoping. The EU AI Act adds real complexity for device-adjacent AI, and firms that map their tools against it now will avoid scrambling in 2026–27. Above all, harmonisation is the story: build once to the shared principles, and you are building for the world’s two largest medicines markets simultaneously.
FAQs
Is there a specific law regulating AI in drug development?
No single dedicated law. AI is governed by a layered mix of existing quality rules (GxP, 21 CFR Part 11, EU GMP Annex 11), new AI-specific guidance from the FDA and EMA, and — in the EU — the horizontal EU AI Act.
What is the FDA’s AI guidance?
In January 2025 the FDA published draft guidance proposing a seven-step, risk-based credibility-assessment framework for AI used to support regulatory decisions on drugs and biologics, with final guidance expected in 2026.
What is the EMA’s position on AI?
The EMA finalised a reflection paper in September 2024 setting out a risk-based, lifecycle approach to AI across the medicines lifecycle. It is non-binding but signals the agency’s expectations, and companies can seek scientific advice on AI methods.
What are the FDA–EMA guiding principles?
In January 2026 the FDA and EMA jointly issued ten Guiding Principles of Good AI Practice in Drug Development, covering ethical values, risk-based approaches, context of use, data governance, human oversight, lifecycle management and transparency.
Does the EU AI Act apply to pharma?
Yes, where relevant. AI that is or is embedded in a medical device or IVD is automatically high-risk under the EU AI Act, with high-risk obligations applying from August 2026 (extended to August 2027 for AI in regulated products).
What does “context of use” mean?
Context of use is the specific role an AI model plays in a decision — what question it answers and how much the decision relies on it. Regulators expect the depth of validation to match the model’s context of use and risk.
Editor’s Note. AI regulation in drug development is moving quickly. This guide reflects the state of the rules as of 2026 — including the FDA’s pending final guidance and the EU AI Act’s phased application — and will be updated by the LSGN editorial team as major developments emerge.
Related Content
- Foundational read: AI in Drug Discovery and Development: The Complete Guide (pillar).
- Closely related: GxP-Compliant AI: Validating Machine Learning for Regulated Drug Development.
- Related: AlphaFold Explained: How AI Protein Structure Prediction Changed Drug Discovery.


